Em mais uma edição do ‘Drops de Genômica’, o oncologista André Murad explica as tecnologias para análise do perfil molecular tumoral de célula única.
Em mais uma edição do ‘Drops de Genômica’, o oncologista André Murad explica as tecnologias para análise do perfil molecular tumoral de célula única.
Por André Marcio Murad*
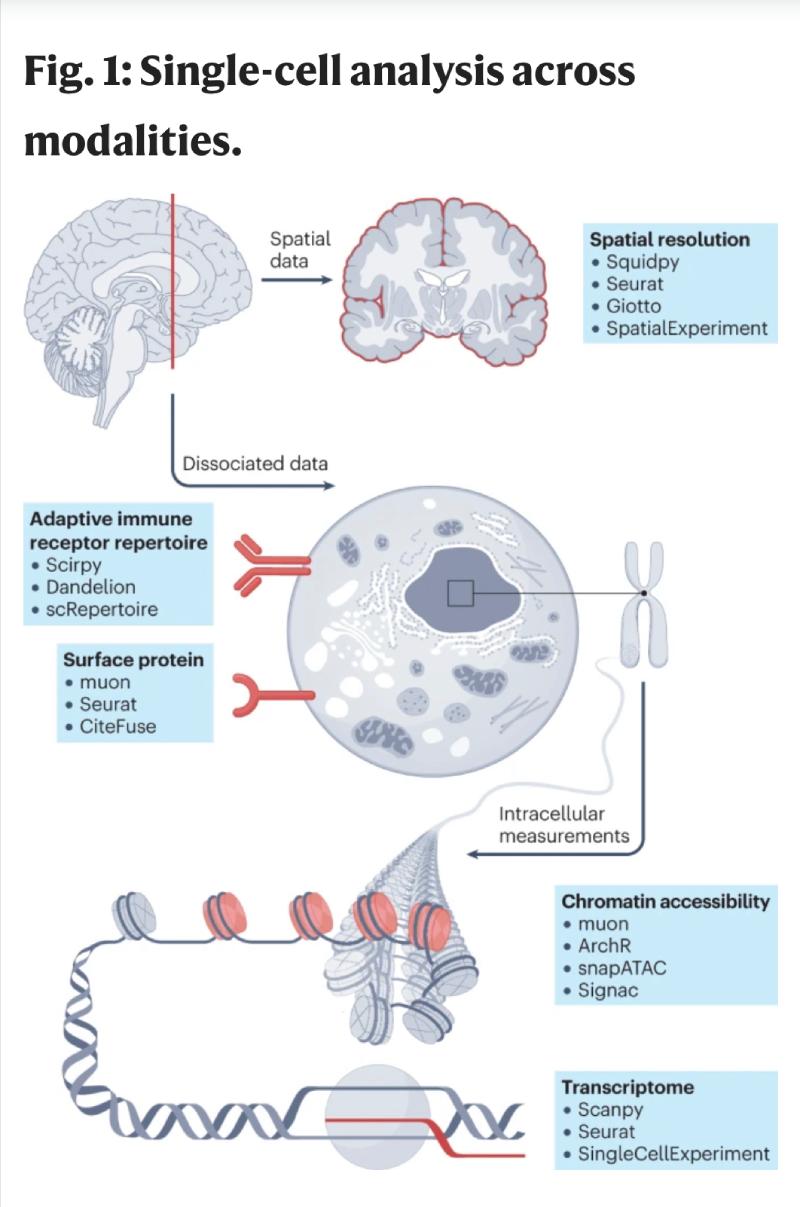
Avanços recentes nas tecnologias de célula única permitiram uma avaliação mais precisa do perfil molecular de alto rendimento de células de variadas modalidades e origens. Os dados transcriptômicos de célula única agora podem ser complementados pela acessibilidade da cromatina, expressão de proteínas de superfície, perfis de repertório de receptores imunes adaptativos e informações espaciais.
A crescente disponibilidade de dados de célula única em todas as modalidades motivou o desenvolvimento de novos métodos computacionais para ajudar os analistas a obter análises biológicas mais complexas. À medida que o campo cresce, torna-se cada vez mais difícil navegar no vasto cenário de ferramentas e etapas de análise.
Transcriptoma
As tecnologias de sequenciamento de RNA de célula única (scRNA-seq) revolucionaram a biologia molecular, permitindo a medição de perfis de transcriptoma em escala e resolução sem precedentes. Avanços na tecnologia experimental motivaram inovações em larga escala em métodos computacionais, levando a mais de 1.400 ferramentas atualmente disponíveis para analisar dados scRNA-seq. Estruturas computacionais e repositórios de software, como Bioconductor, Seurat e Scanpy, complementados por benchmarks de métodos e fluxos de trabalho de melhores práticas, permitiram que analistas de dados navegassem nesse espaço e construíssem canais de análise. Essa interação de inovação experimental e computacional tem permitido muitas descobertas de marcos biológicos que revelam a heterogeneidade celular do tecido.
No entanto, o scRNA-seq captura apenas uma camada da complexa maquinaria regulatória que governa a função celular e a sinalização. Para complementar isso, esforços consideráveis foram feitos para medir outras modalidades na resolução de uma única célula, incluindo acessibilidade da cromatina, proteínas de superfície, repertórios de receptores de células T (TCR)/receptores de células B (BCR) e localização espacial, permitindo a descoberta de inúmeras assinaturas reguladoras de vários tipos de câncer, além do diabetes mellitus, resposta desregulada do sistema imunológico inato e adaptativo contra o coronavírus 2 da síndrome respiratória aguda grave (SARS-CoV-2) e melhor compreensão dos efeitos imunossupressores do microambiente tumoral na resolução espacial. A inovação experimental levou ao desenvolvimento de várias inovadoras ferramentas computacionais para inúmeras modalidades de célula única.
Entretanto, deve-se ressaltar que a falta de fluxos de trabalho de melhores práticas torna desafiadora a navegação no vasto cenário de novas ferramentas. Além disso, embora as melhores práticas computacionais e recomendações de ferramentas tenham sido descritas anteriormente para scRNA-seq, elas podem estar desatualizadas ou incompletas.
Acessibilidade da cromatina
A análise de elementos regulatórios é essencial para decifrar a diversidade celular e entender a tomada de decisão celular. A expressão gênica é controlada por uma complexa interação de mecanismos regulatórios, incluindo epigenética e acessibilidade da cromatina. Para obter informações sobre a dinâmica do estado da cromatina no nível de célula única, o ensaio de célula única para sequenciamento de cromatina acessível por transposase (scATAC-seq) mede a acessibilidade da cromatina em todo o genoma em células individuais.
Expressão de proteína de superfície
A transcrição e a acessibilidade da cromatina são proxies para o estado celular, atividade e regulação. Os produtos reais gerados, as proteínas, assumem tarefas intracelulares ou extracelulares, e um subconjunto de proteínas é apresentado na superfície celular. A expressão de proteínas de superfície auxilia na identificação de tipos de células, como as células hematopoiéticas do sistema imunológico, cuja anotação é baseada em marcadores que geralmente são usados em experimentos de citometria de fluxo ou citometria de massa. Eles podem ser usados posteriormente para validar genes geneticamente eliminados específicos usando, por exemplo, o pipeline Mixscape.
Os protocolos mais amplamente utilizados para scRNA-seq combinado e perfis de proteínas de superfície são o CITE-seq e o REAP-seq, com a principal diferença sendo as etiquetas derivadas de anticorpos (ADTs) que são usadas para quantificar os níveis de expressão de proteínas de superfície.