
 Ainda existem lacunas em nosso conhecimento sobre causas hereditárias e esporádicas de malignidades hematológicas (MH) e insuficiência da medula óssea (IMO) que impedem o diagnóstico ideal, a vigilância da doença e o tratamento. Estudo destacado no ASH 2023 relatou a descoberta do ERG como um novo gene de predisposição para IMO e MH.
Ainda existem lacunas em nosso conhecimento sobre causas hereditárias e esporádicas de malignidades hematológicas (MH) e insuficiência da medula óssea (IMO) que impedem o diagnóstico ideal, a vigilância da doença e o tratamento. Estudo destacado no ASH 2023 relatou a descoberta do ERG como um novo gene de predisposição para IMO e MH.
ERG é um oncogene conhecido, normalmente por meio de fusões de genes, levando à superexpressão desregulada de ERG em cânceres sanguíneos e sólidos. Neste estudo multicêntrico australiano, os pesquisadores identificaram uma variante germinativa do domínio ERG ETS p.Y373C segregando com trombocitopenia em uma mãe que progrediu para Leucemia Mieloide Aguda (27 anos) e depois para Síndrome Mielodisplásica (SMD) relacionada à terapia (35 anos), e em seus 2 filhos. Todos os três mostraram perda neutra de cópia de heterozigosidade de todo ou parte do cromossomo 21q, incluindo o locus ERG, com o filho mais velho apresentando pelo menos 2 eventos de resgate genético somático (RGS).
Os pesquisadores descrevem que a possibilidade de variantes causais de RUNX1 foi descartada, com o menor evento somático cnLOH começando no gene RUNX1, mas não abrangendo o domínio RUNT, onde a maioria das variantes patogênicas missense estão localizadas. ERG, um gene altamente restrito (LOEUF <0,33), é crítico para a hematopoiese definitiva, a função das células-tronco hematopoiéticas adultas (HSC) e a manutenção das plaquetas. Uma variante de linha germinativa heterozigótica correspondente idêntica (p.Y343C) no gene mais próximo do ERG por homologia, FLI1, causa distúrbio hemorrágico do tipo plaquetário-21 (BDPLT21, OMIM #617443).
“Através de colaborações globais, identificamos 15 variantes heterozigóticas no gene ERG, 13 das quais são variantes missense e 2 variantes truncadas, em 17 indivíduos com citopenia e/ou MH (principalmente mieloide) ou linfedema”, esclarecem. O início dos sintomas hematológicos variou desde o nascimento até 38 anos para variantes truncadas e restritas do domínio ETS. Destas 15 variantes, 12 foram confirmadas como linha germinativa, incluindo 2 de novo. Apenas 4 transmissões meióticas foram observadas. Nenhuma das variantes missense no domínio ETS altamente conservado do ERG, que medeia a ligação ao DNA, as interações proteína-proteína e a localização nuclear, está presente no gnomAD.
O estudo caracterizou funcionalmente 19 variantes de ERG, 12 potencialmente patogênicas, 1 variante patogênica de camundongo conhecida e 3 controles populacionais, demonstrando que a maioria das variantes missense do domínio ETS apresentam características de perda de função, interrompendo a transativação transcricional (Figura), ligação ao DNA e/ ou localização nuclear in vitro. Dados preliminares robustos de modelos ex vivo de superexpressão de ERG em células hepáticas fetais de camundongos em cultura de tecidos (independência de citocinas), um ensaio de transplante de camundongo e modelos anteriores de camundongos ERG com mutação de linha germinativa são concordantes com variantes missense do domínio ETS, sendo a perda de função (LOF) comparada ao ERG de tipo selvagem e controles benignos. Juntos, esses dados fornecem estudos funcionais clínicos, in vitro e ex vivo, implicando variantes de LOF na predisposição a doenças hematológicas. Mutações LOF ERG também ocorrem em casos esporádicos de MH.
A análise mostra que, como parte de um estudo populacional do Genomics England Research Consortium, 4 variantes ERG truncadas foram descritas recentemente em 7 indivíduos em 4 famílias, com 3 transmissões meióticas e um caso de novo com linfedema primário. Agora, os pesquisadores australianos adicionam duas novas variantes missense. Um paciente apresentou RGS através do locus ERG no sangue. Os fenótipos sanguíneos não foram descritos. Esses resultados demonstram que as variantes germinativas do ERG predispõem a diversas citopenias, IMO e MH em crianças e adultos. Na família mencionada acima, a mãe recebeu um aloHSCT não relacionado devido a t-SMD, enquanto seus 2 filhos com citopenias continuam sendo monitorados.
Os autores descrevem que a síndrome ERG é paralela à síndrome de deficiência de GATA2 (HM e linfedema) e ao distúrbio plaquetário familiar RUNX1 - malignidade mieloide (trombocitopenia e MH). Assim como os conhecidos genes de doenças GATA2 e RUNX1, o ERG também é membro do fator de transcrição envolvido na manutenção e diferenciação do HSC. O ERG acrescenta-se a uma lista crescente de genes cuja expressão desregulada contribui para MH e outros tipos de câncer.
“A identificação de variantes ERG da linha germinal, como as descritas neste estudo, tem implicações clínicas diretas para o manejo do paciente e da família, incluindo diagnóstico, aconselhamento, vigilância e estratégias de tratamento, como seleção de doadores para transplante de medula óssea e potencial para terapias alvo, incluindo terapia genética e celular”, conclui a análise.
A história natural desta nova síndrome exigirá a identificação cuidadosa das lesões germinativas, sendo necessários estudos longitudinais adicionais em mais pacientes e famílias.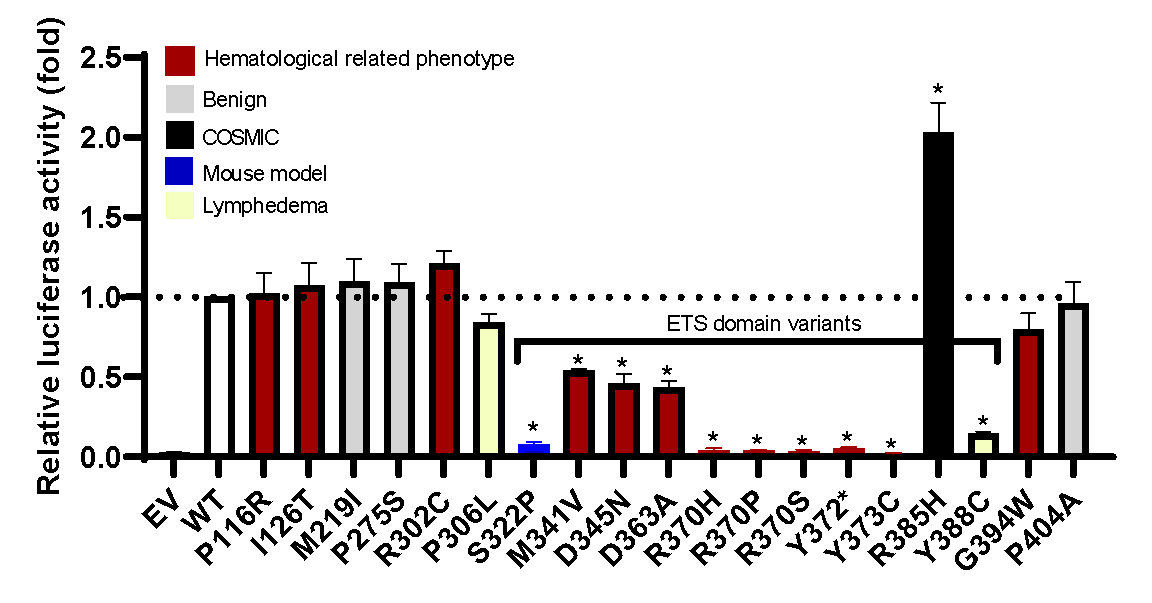
Referência: LBA-6 ERG Is a New Predisposition Gene for Bone Marrow Failure and Hematological Malignancy - Hamish S Scott et al.

